A importância dos lisossomos para a nossa saúde
Autores
Stefanie Flunkert, Tatjana Hirschmugl
Jovens revisores
Programa de extensão da Gleeson College, Siddharth

Resumo
Os lisossomos são estruturas celulares especializadas que servem como um sistema de coleta de lixo das células do corpo. Eles contêm componentes que limpam diversos tipos de “lixo” celular e são responsáveis por ativar o processo de reciclagem. Quando os lisossomos não funcionam corretamente, o lixo se acumula nas células, levando eventualmente à doença e à morte celular. Isto pode levar ao desenvolvimento de várias doenças de armazenamento lisossomal. Os sintomas destas doenças variam em gravidade, de sintomas que quase não afetam a vida do paciente até sintomas que já começam no nascimento e que reduzem significativamente a expectativa de vida. Duas opções de tratamento estão disponíveis para algumas dessas doenças que podem melhorar os sintomas: a terapia de reposição enzimática e a terapia de redução de substrato. Uma nova abordagem de pesquisa chamada terapia genética oferece uma cura potencial. Neste artigo explicaremos o papel dos lisossomos e o que acontece quando eles não funcionam corretamente. Também forneceremos detalhes sobre os tratamentos disponíveis e como a terapia genética pode ser um avanço neste campo.
O que são lisossomos?
Lisossomos são estruturas especializadas dentro das células, cada uma envolvida por uma membrana (exibida nas Figuras 1c e 1d como corte). Eles contêm muitos tipos de enzimas (exibidas em detalhes na Figura 1c), que são proteínas especializadas necessárias para cumprir as funções dos lisossomos. Os lisossomos podem ser encontrados em quase todos os tipos de células animais, incluindo células humanas. Em cada célula, existem de 50 a 1.000 lisossomos. Quando os pesquisadores começaram a analisar a função dos lisossomos, pensaram que fosse apenas um sistema de coleta de lixo das células. Eles observaram que os lisossomos absorvem todo o lixo celular e o digerem, com a ajuda de enzimas, como se fosse uma central de reciclagem que tritura o lixo antes de utilizá-lo na fabricação de novos materiais (Figuras 1a, 1c).
Agora se sabe que a função dos lisossomos é muito mais complexa. Eles também fornecem lixo para outros centros de reciclagem e estão envolvidos na reparação de membranas celulares danificadas. Ainda mais importante, os lisossomos funcionam como sensores (Figuras 1a, 1c) que podem dizer se uma célula está saudável, se faltam nutrientes ou se foi atacada por bactérias ou vírus. Uma vez que o lisossomo detecta tal problema, pode tomar medidas apropriadas para melhorar o bem-estar da célula (exibido como uma antena que detecta o cheiro nas Figuras 1a, 1c). Pode, por exemplo, informar o sistema imunológico sobre intrusos, para que as defesas do corpo possam tornar-se ativas e lutar contra invasores perigosos. Portanto, os lisossomos também são importantes vigilantes, mantendo as células saudáveis [1].
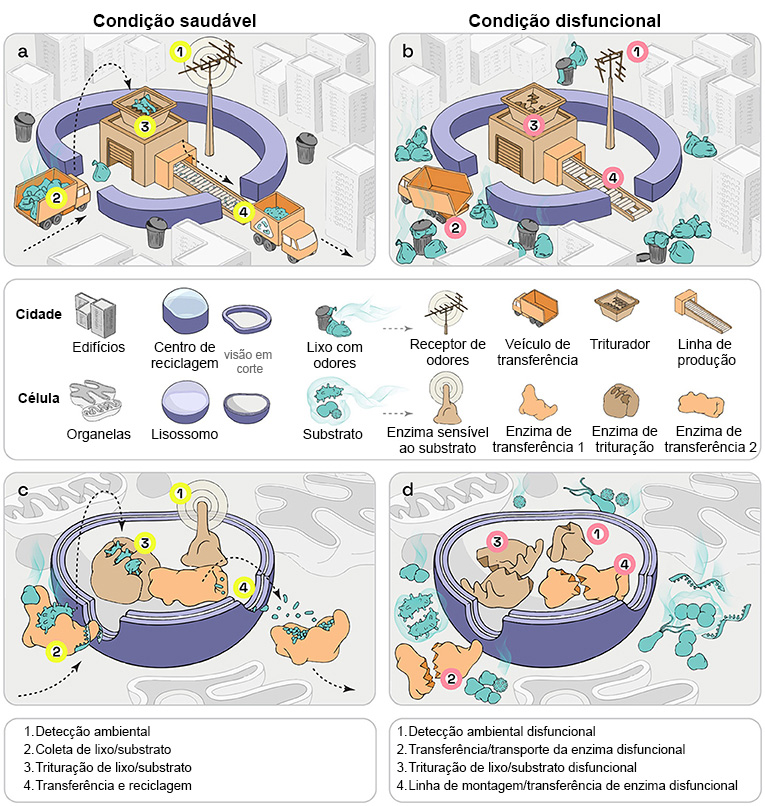
O lisossomo saudável (a) e o disfuncional (b) são apresentados como coletor de lixo e centro de reciclagem. (c, d) Funções lisossômicas em nível celular. (a, c) Na situação saudável, o lisossomo pode sentir o ambiente da célula (1). Além disso, coleta o lixo (2), decompõe (3) e recicla-o (4). Nas doenças de armazenamento lisossômico [condição disfuncional, (b, d)], uma dessas funções não está atuando. Por exemplo, o sensor (1), o coletor (2), o sistema de trituração (3) ou o sistema de reciclagem (4) podem estar quebrados. Nestes casos, o lixo celular acumula-se até as células adoecerem.
As doenças de armazenamento lisossômico
O que acontece quando os lisossomos não funcionam como deveriam? Os distúrbios do sistema lisossômico podem ter muitas causas, pois o funcionamento adequado dele envolve mais de 60 enzimas.
Em uma doença de armazenamento lisossômico, uma das enzimas ou proteínas que são importantes para o funcionamento do lisossomo não funciona corretamente. Esse defeito causa um acúmulo de lixo que a enzima deveria reciclar (Figuras 1b, 1d). Imagine os catadores de lixo do seu bairro retirando todo o lixo que você separou e colocou do lado de fora para coleta, exceto o lixo plástico. Depois de algumas semanas, as ruas se encheriam de lixo plástico. É exatamente isso que acontece nas células doentes: o lixo celular que já não pode ser reciclado obstrui a célula e acaba por matá-la.
Os cientistas chamam esse lixo de substrato celular, que não pode mais ser reciclado e, portanto, obstrui a célula e eventualmente a mata. (Figuras 1b, d). Como cada uma das enzimas realiza uma tarefa separada, a célula – e consequentemente o paciente – enfrenta problemas específicos dependendo de qual enzima não funciona adequadamente. Até agora, são conhecidas mais de 60 doenças de armazenamento lisossômico, que podem afetar várias partes do corpo, como esqueleto, cérebro, pele ou coração, dependendo da enzima afetada.
As doenças de depósito lisossômico pertencem ao grupo das doenças raras. Uma doença é considerada rara se não afetar mais de 1 em cada 1.500 pessoas. No seu conjunto, todas as doenças de depósito lisossômico afetam cerca de 1 em cada 4.000 pessoas. Devido à sua raridade, os médicos raramente atendem pacientes com doença de armazenamento lisossômico, por isso não têm muita experiência com eles. Isto significa que os pacientes com doenças de armazenamento lisossômico podem lutar durante anos para obter um diagnóstico correto.
A maioria das doenças de depósito lisossômico é grave, e resulta em morte precoce. Algumas crianças morrem logo após o nascimento, enquanto outros pacientes apresentam os primeiros sintomas durante a idade adulta [2]. Muitas doenças de armazenamento lisossômico recebem o nome do(s) médico(s) que as descreveram pela primeira vez, como a doença de Krabbe, Gaucher, Niemann-Pick, Hunter, Hurler e Tay-Sachs. Além disso, todas essas doenças são nomeadas com base na proteína defeituosa: mucopolissacaridose, mucolipidose, gangliosidose, esfingolipidose e muitas mais. As doenças nomeadas em homenagem aos médicos têm, portanto, dois nomes: o nome comum baseado no médico, mais o nome baseado no defeito proteico defeituoso.
O Caso de Hannah
Apenas dois dias depois do nascimento de Hannah, os médicos perceberam que algo estava errado, pois o baço dela estava maior que o normal. Os médicos levaram cinco meses para encontrar o diagnóstico correto, e Hannah foi diagnosticada com uma forma grave da doença de Gaucher que também afeta o cérebro. Os médicos lhe deram nove meses de vida. Durante os meses seguintes, a coordenação motora de Hannah sofreu e o seu desenvolvimento atrasou bastante. Além do baço, seu fígado foi afetado e ela não conseguia mais sentar-se sem ajuda ou pegar coisas sozinha. Embora parecesse ser uma criança feliz, seu cérebro acabou sendo destruído pela doença. Hannah viveu 3 anos antes de falecer nos braços dos pais. Se você quiser saber mais sobre Hannah, visite este site.
Existe tratamento ou cura?
Como a maioria das doenças de depósito lisossômico apresentam sintomas graves, os investigadores procuram urgentemente uma cura. Até agora, as terapias disponíveis só podem melhorar os sintomas: não é possível uma cura completa. Existem algumas opções terapêuticas que já são utilizadas para algumas dessas doenças ou que estão atualmente em desenvolvimento. Esta seção descreverá essas opções e seus desafios.
Terapia de reposição enzimática
Durante a terapia de reposição enzimática, o paciente é tratado com a enzima que não é produzida pelas próprias células. Como uma doença de armazenamento lisossômico pode ser causada pela falta de apenas uma das muitas enzimas lisossomais, cada uma destas doenças necessita do seu próprio medicamento de substituição enzimática (Figura 2a). Ao fornecer ao corpo a enzima que falta, o lisossomo pode funcionar adequadamente, evitando o acúmulo de lixo celular, o substrato.
A terapia de reposição enzimática pode manter as células funcionais e saudáveis. Para um tratamento bem-sucedido, as terapias de reposição enzimática devem ser iniciadas o mais cedo possível para evitar acúmulo intenso de substrato. Como as enzimas se decompõem rapidamente no corpo, elas devem ser injetadas regularmente pelo resto da vida do paciente.
Outro problema com as injeções de enzimas é que as enzimas não conseguem atingir o cérebro devido à barreira hematoencefálica, que protege o cérebro de tudo que possa potencialmente prejudicá-lo. Esta barreira bloqueia a entrada de enzimas no cérebro, pois são muito grandes (Figura 2a). Como muitas doenças de armazenamento lisossômico são causadas pelo mau funcionamento de enzimas que também são importantes para o funcionamento adequado do cérebro, a terapia de reposição enzimática tem apenas um benefício limitado para esses pacientes [3].
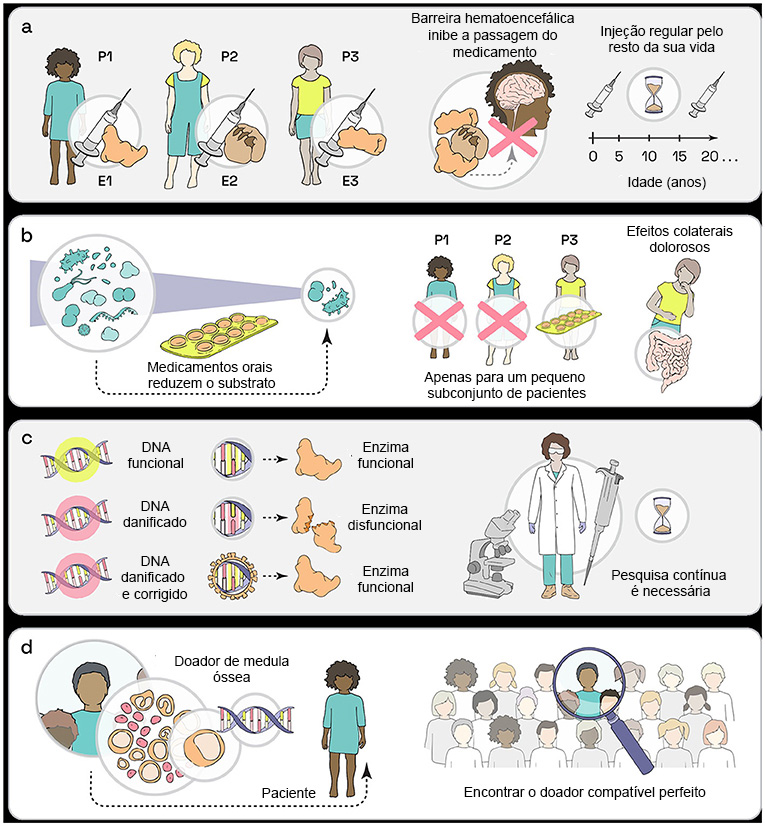
(a) A reposição enzimática envolve injeções regulares da enzima, mas ela não consegue chegar ao cérebro. (b) Na redução de substrato, os pacientes podem tomar comprimidos, mas isso geralmente traz fortes efeitos colaterais. (c) A terapia genética tenta corrigir a razão subjacente de uma doença e pode ser capaz de fornecer uma cura. (d) A doação de medula óssea já é utilizada, mas é difícil encontrar um doador perfeitamente compatível (P, paciente; E, enzima).
Terapia de redução de substrato
Uma abordagem alternativa para melhorar a vida dos pacientes é a terapia de redução de substrato, tratamento que utiliza medicamentos que reduzem a produção do substrato. Isto significa que, em vez de limpar o lixo, a terapia de redução de substrato minimiza o acúmulo de lixo. Portanto, se o seu sistema de coleta de lixo não funciona para lixo plástico, produza o mínimo possível de lixo plástico! Uma grande vantagem da terapia de redução de substrato é que ela é tomada por via oral na forma de comprimido, embora esteja atualmente disponível apenas para algumas doenças de armazenamento lisossômico (Figura 2b). Por outro lado, as grandes desvantagens destas terapias são os fortes efeitos colaterais que muitos pacientes desenvolvem, como diarreia e problemas estomacais [4].
Terapia Gênica
As doenças de armazenamento lisossômico são causadas principalmente por um único defeito no gene de uma enzima lisossômica; é como um erro no plano de construção da proteína. Uma nova abordagem terapêutica visa fixar o gene subjacente para que as células possam produzir a enzima e reciclar o lixo novamente.
Embora sejam necessárias enzimas que funcionem adequadamente em todo o corpo, resgatar a enzima que funciona inadequadamente em algumas células do paciente pode produzir enzima suficiente para prevenir os sintomas da doença ou mantê-los no mínimo. Tratar uma doença fixando um gene, o DNA, é chamado de terapia gênica, que visa consertar um gene defeituoso que de outra forma causaria uma doença.
Essa terapia precisaria ser realizada individualmente para cada paciente, pois os pacientes provavelmente apresentam defeitos genéticos únicos (Figura 2c). Atualmente, os pesquisadores buscam o melhor método para corrigir os genes dos pacientes. Por exemplo, eles tentam usar vírus modificados para introduzir com segurança o gene saudável nas células dos pacientes. Experimentos que exploram esta terapia são realizados em células doadas de pacientes com doença de armazenamento lisossômico. Todos os experimentos são realizados fora do corpo, para garantir que nenhum paciente seja ferido.
Doação de medula óssea e células sanguíneas
Uma alternativa já estabelecida à terapia genética é o tratamento de pacientes com doença de armazenamento lisossômico com medula óssea ou células sanguíneas de um doador humano saudável. Isto é semelhante a um transplante de rim ou fígado. O problema com este método é que a pessoa doadora que fornece voluntariamente algumas células ou tecidos saudáveis do seu corpo precisa ter uma combinação biológica perfeita com o paciente, o que é difícil de encontrar (Figura 2d). Embora essas células novas e saudáveis muitas vezes produzam enzimas suficientes para prevenir grandes danos aos órgãos, ainda não é suficiente para o paciente estar livre de sintomas [5].
Resumo
Embora as doenças de depósito lisossômico sejam muito raras, seus sintomas podem ser variados e graves. Atualmente, não existem opções de tratamento perfeitas disponíveis, mas os pesquisadores continuam explorando as possibilidades. Especificamente, espera-se que a abordagem da terapia genética resulte em opções de tratamento individualizadas e talvez até na cura para pacientes com doença de armazenamento lisossômico. Enquanto isso, os pacientes podem ser ajudados com uma variedade de medicamentos que devem ser escolhidos para cada paciente individualmente.
Glossário
Lisossomo: Estrutura celular especializada, rodeada por uma membrana, que serve como sistema de coleta de lixo das células do corpo.
Enzimas: Tipo específico de proteína que pode acelerar uma reação química no corpo.
Doença de armazenamento lisossômico: Doenças causadas por lisossomos que não funcionam adequadamente.
Substrato: Lixo celular que não pode mais ser reciclado e, portanto, obstrui a célula e eventualmente a mata.
Terapia de reposição enzimática: Tratamento com uma enzima que restaura a função de uma enzima que está faltando no corpo.
Terapia de Redução de Substrato: Tratamento que reduz a produção de substrato.
Terapia Gênica: Conserta um gene defeituoso que causa uma doença.
Doador: Uma pessoa que fornece voluntariamente algumas células ou tecidos saudáveis do seu corpo
Referências
[1] Inpanathan, S., e Botelho, R. J. 2019. The lysosome signaling platform: adapting with the times. Front. Cell Dev. Biol. 7, 113. doi: 10.3389/fcell.2019.00113
[2] de O. Poswar, F., Vairo, F., Burin, M., Michelin-Tirelli, K., Brusius-Facchin, A. C., Kubaski, F., et al. 2019. Lysosomal diseases: overview on current diagnosis and treatment. Genet. Mol. Biol. 42, 165–177. doi: 10.1590/1678-4685-gmb-2018-0159
[3] Fernández-Pereira, C., Millán-Tejado, B. S., Gallardo-Gómez, M., Pérez-Márquez, T., Alves-Viller, M., Melcon-Crespo, C., et al. 2021. Therapeutic approaches in lysosomal storage diseases. Biomolecules 11, 1775. doi: 10.3390/biom11121775
[4] Coutinho, M., Santos, J., Matos, L., e Alves, S. 2016. Genetic substrate reduction therapy: a promising approach for lysosomal storage disorders. Diseases 4, 33. doi: 10.3390/diseases4040033
[5] Nagree, M. S., Scalia, S., McKillop, W. M., e Medin, J. A. 2019. An update on gene therapy for lysosomal storage disorders. Exper. Opini. Biol. Ther. 19, 655–670. doi: 10.1080/14712598.2019.1607837
Citação
Flunkert S e Hirschmugl T (2023) How Lysosomes Keep Us Healthy. Front. Young Minds. 11:1109280. doi: 10.3389/frym.2023.1109280
Este é um artigo de acesso aberto distribuído sob os termos da Creative Commons Attribution License (CC BY). O uso, distribuição ou reprodução em outros fóruns é permitido, desde que o(s) autor(es) original(is) e o(s) proprietário(s) dos direitos autorais sejam creditados e que a publicação original nesta revista seja citada, de acordo com a prática acadêmica aceita. Não é permitido nenhum uso, distribuição ou reprodução que não esteja em conformidade com estes termos.
Encontrou alguma informação errada neste texto?
Entre em contato conosco pelo e-mail:
parajovens@unesp.br


